The pharma industry from Paul Janssen to today: why drugs got harder to develop and what we can do about it
In 1953, aged 27, Paul Janssen set up the research laboratory on the third floor of his parents’ Belgian drug import firm from where he would grow his eponymous pharmaceutical company. In the years between the 50s and 90s when he was most active, Janssen and his team developed over 70 new medicines, many of which are still in use today. Such prolificacy is unlikely to be repeated any time soon; if current trends hold, a drug discovery scientist starting their career today is likely to retire without ever having worked on a single drug that makes it to market.

The cost to discover and develop a drug today is orders of magnitude higher than in the 1950s. Despite this, the probability that a drug entering clinical trials will eventually reach the market has hardly improved in the intervening years. If Janssen were born today, there’s little chance he would be able to repeat his success. He would probably not even get the chance to start.
What changed? Some lay the blame for these deteriorating conditions on regulators like the FDA, claiming that if we were to abolish regulators we would release the stranglehold on industry and unleash a deluge of stalled medicines. Others blame ‘big pharma’, claiming the industry is suppressing cures — more interested in price gouging on old drugs than investing in R&D. These explanations lack nuance. In reality, the productivity crisis in the pharmaceutical industry is the culmination of decades of just about every aspect of drug discovery and development getting gradually harder and more expensive.
So how did one man and his start-up manage to achieve a level of output that would be the envy of today’s pharmaceutical giants?
Act 1: Paul Janssen, and the rise of ‘traditional’ drug discovery and development
Before Janssen, most pharmaceutical preparations were natural products or combinations of known compounds. During the ‘Golden Age’ of antibiotic discovery, beginning in the 1940s and peaking in the mid 50s, new antibiotic classes were being discovered on a near-yearly basis. Soil microorganisms were a particularly rich source of antibiotics. Chloramphenicol, for instance, was identified from samples of Streptomyces bacteria collected from a mulched field by Parke-Davis company scientists on a field expedition to Venezuela.
Natural products are enriched for biologically active compounds, and nature remains a useful source of new drugs even today. That said, the industry’s historical focus on natural products limited the drugs that could be discovered to those that could be found serendipitously out in the world. Advances in synthetic organic chemistry unlocked the ability to systematically create drugs on the laboratory benchtop. This vastly expanded the space of possible drug molecules that could be probed, and enabled the industrialisation of the drug discovery process.
Although artificial chemical synthesis arose in the mid 1800s, and structurally simple drugs like the barbiturate sedatives and ‘miracle’ sulfonamides — the first broadly effective antibiotics — were synthesised in the early 1900s, it was not until the mid-20th century that chemical synthesis and analysis techniques matured enough to be widely useful for industrial drug discovery. Janssen’s ambition was to apply the burgeoning synthetic organic chemistry toolbox to discover and synthesise entirely new drugs.
Chemical space is practically infinite, but useful drugs are a miniscule subset of all possible chemical structures; there are simply too few ways for compounds to interact usefully with living systems to generate chemicals at random and expect them to be useful drugs1. Constrained by limited money and resources, Janssen needed a quick, easy, and cheap discovery methodology — in his words:
It was a matter of life or death — a matter of survival. I don’t believe we really had a clear-cut strategy. We were simply doing whatever we could, and there weren’t many things that we could do then. We didn’t have much money and there were not many researchers. We had to make a lot of simple compounds as quickly and possible and screen them using very simple methods.
One of Janssen’s procedural innovations was a structured process for synthesising and testing new drugs for biological activity. Janssen employed a relatively simple process of swapping molecular building blocks around easily-modifiable central cores of molecules with known activity. With this mix-and-match approach, Janssen’s team quickly iterated and built up a library of standardised components. Janssen had stumbled into virgin territory, and in those days useful drugs were relatively easy to come by: Janssen found a commercialisable drug on his fifth try with ambucetamide, a treatment for muscle spasms. Within a year of starting out, Janssen and his team had synthesised around 500 new compounds.
Five years into the venture, in 1958, Janssen’s team discovered haloperidol. Haloperidol was a fortuitous byproduct of a project that aimed to develop more effective and less addictive painkillers by tweaking pethidine, a synthetic painkiller similar to morphine. In the process of iterating on pethidine, Janssen’s team observed that mice given one of their newly synthesised compounds became unexpectedly calm, sedated, and rigid — showing little of the anticipated agitation. This was an unexpected combination of effects for a pethidine derivative, reminiscent of the neuroleptic antipsychotics like chlorpromazine that had recently come to market; Janssen’s team had stumbled upon a potential new class of antipsychotic treatments for schizophrenia and related disorders.
After this discovery, the project team shifted their focus towards optimising the compound’s antipsychotic properties through subtle structural tweaks, eventually settling on haloperidol as the candidate to be trialled in humans. By 1959, about a year after it was first synthesised, and after a short period of clinical tests in psychiatric wards, haloperidol became widely available in Belgium. Most of Western Europe followed within two years.
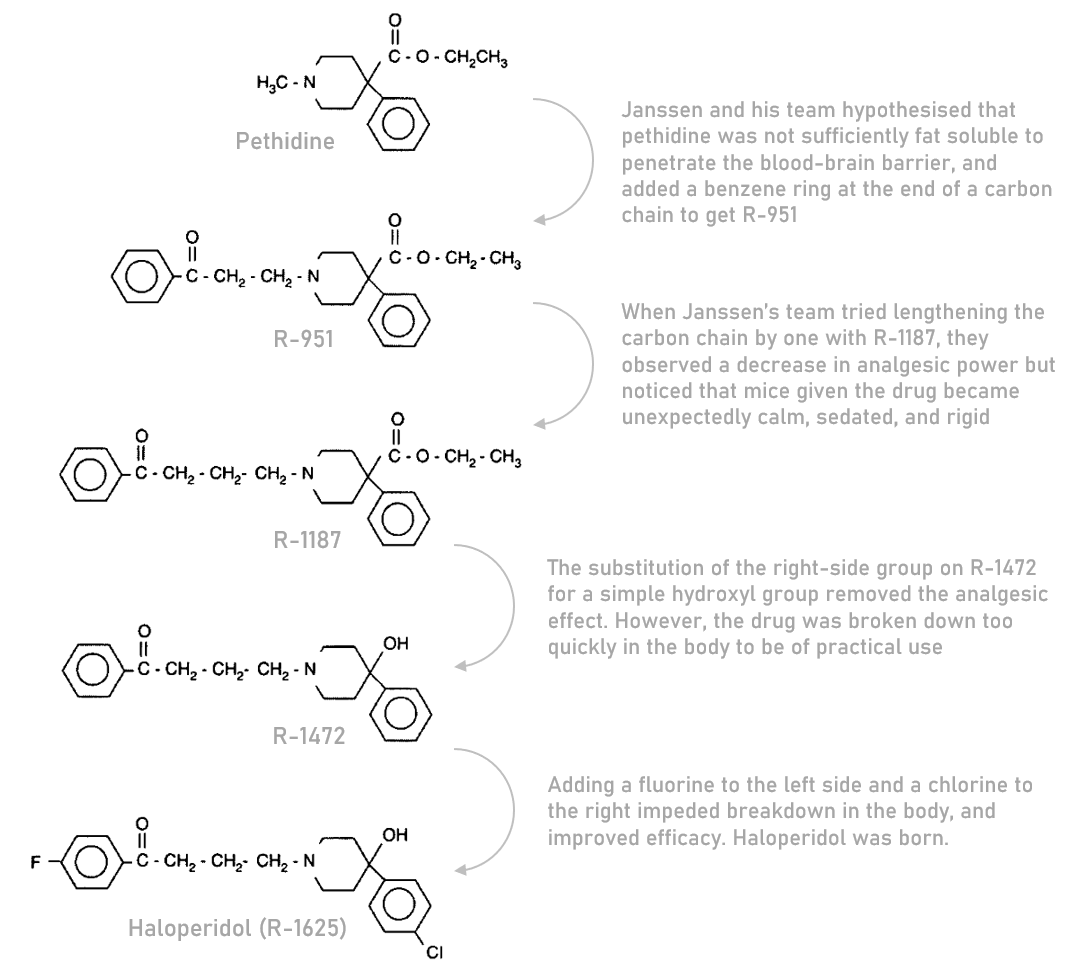
Prior to the 1950s, there was no effective pharmaceutical treatment for schizophrenia. The discovery of multiple ‘first-generation’ antipsychotics in the 50s, including haloperidol, brought about a revolution in psychiatry and made it possible for people with schizophrenia to reintegrate into society. With fewer sedating side effects than chlorpromazine — the first antipsychotic on the market — haloperidol gained rapid adoption among psychiatrists. Although the ‘second-generation’ of antipsychotics discovered in the 80s have fewer side effects and mostly supplanted haloperidol, doctors still prescribe the drug today.
Act 2: How drugs are discovered and developed today
The rapid discovery and real-world application of haloperidol would be nigh-impossible to replicate today. Janssen arrived early to a new field, before there was strong competition, before much of the low-hanging fruit was picked, and before it was widely regulated. In the years since, regulation has been layered on, ossifying the process of drug discovery. We have settled into a drug discovery and development process that is long, inefficient, expensive, and likely to fail.
On the path to patients, candidate medicines must successfully navigate a gauntlet of experiments, trials, and regulation that typically runs for about a decade from end-to-end. The standard process to bring a drug to market today can be broken down into four sequential stages: drug discovery, preclinical development, clinical trials, and regulatory approval.
Basic research is upstream of drug discovery and contributes by enriching our fundamental understanding of disease biology and finding new drug ‘targets’. Most basic research takes place in academia, but drug companies (of all sizes) do some as well. Basic research helps discover ‘targets’ — biological entities, most often proteins, thought to have an effect on a particular disease when modulated by the binding of a drug molecule. Janssen didn’t know it at the time, but haloperidol exerts its effects by blocking dopamine receptors on neurons in the brain. Dopamine receptors are now a well established target in schizophrenia.
The starting point for drug discovery is usually some sort of screen: running hundreds, thousands, or even millions of compounds through parallel (high-throughput) experiments to see which, if any, usefully affect a target of interest. These could be simple binding assays, where researchers put a purified protein target in microwell plates (plastic rectangles with huge numbers of tiny holes to do the tests in), pipette compounds among the wells, and check whether any of them stick to the target.
An alternative approach is phenotypic screening, which assesses the effects of candidate drugs on the outwardly observable traits of living systems — like an individual cell in a petri dish, or a mouse —, rather than pre-specified targets in isolation. Before the reductionist target-first paradigm took hold, most discovery programs relied on phenotypic screens. Janssen’s team used this method to test how mice responded to the pain induced by a searing hot plate after they were given the pethidine derivatives. The experiments that led to the discovery of haloperidol weren’t designed to detect new antipsychotics, but their open-ended nature led Janssen’s team to haloperidol regardless.
Often, molecules that come out of a screen are just starting points for further optimisation work. At first, these starter molecules may bind (i.e. stick to) their target weakly, or have undesirable chemical properties, like poor solubility, that makes them hard to usefully dose in practice, or they may metabolise into toxic ‘breakdown products’ in the body. As Janssen did with haloperidol, chemists make structural tweaks until they arrive at a satisfactory ‘lead candidate’ molecule. The active drug ingredient may need to be combined with other compounds (excipients), like sugars, which make it easier to administer, or which help the body distribute and metabolise the drug. For example, semaglutide (a member of the potent GLP-1 agonist class of diabetes and obesity drugs) is normally injected because if it’s taken orally it just gets broken down or fails to pass through the gut walls. Novo Nordisk developed an oral version of semaglutide by formulating the drug with a permeability enhancer called SNAC. SNAC carries the semaglutide peptide through the walls of the gut and eases its absorption into the bloodstream.
Once a lead candidate has been developed that seems sufficiently promising, it is ushered into preclinical development. The goal is to understand how the drug works in the body, and to prove it’s safe enough for human trials.
Preclinical development means giving the drug to cell cultures (‘in vitro’) and whole animals (‘in vivo’) — commonly rats or mice; sometimes dogs, pigs, or monkeys — and demonstrating the quality and purity of the manufactured drug to a high standard. The end result is an understanding of the risks and side effects associated with the drug, how the drug is metabolised and distributed in the body, and a plausibly safe dose range to take into humans. Often, if problems with the drug are discovered during this phase they will feed back into and inform further refinement of the drug candidate.
After the drug candidate has been cleared for human testing, the clinical phase begins – where drugs are tested in humans for first safety and then efficacy. Typically, phase I trials focus on safety and finding an appropriate dose, often in healthy volunteers; phase II on establishing preliminary evidence of efficacy in patients; phase III on confirming efficacy in a larger sample of patients, and collecting robust safety data.
The most important data collected in a trial is the specific clinical objective known as the ‘primary endpoint’ of the trial2, such as improvements in survival or pain severity scales. A failed trial is one in which the drug misses the target level of performance on the primary endpoint. The primary endpoint and associated statistical analysis plan is specified before the start of the trial — this (mostly) stops drug developers from cherry picking cuts of data in an attempt to artificially make their drugs look better after a trial failure.
Selecting a primary endpoint is a balance between finding an outcome that is meaningful, sensitive enough to detect the effect of a drug (if it has an effect), and can be measured accurately and in a timely fashion within the context of a clinical trial. Before starting a trial, companies will consult with regulators to get tacit approval on their trial design.
The gold standard of evidence for new drugs is the randomised double-blind clinical trial. In this type of trial, new drugs are tested against a control group receiving the ‘standard of care’ — the conventional treatment for the condition in actual practice, or a placebo if no established standard of care exists3. Volunteers are randomly assigned to the new drug group or the control group, and the trial is ‘double-blind’ in that neither the patients nor the clinicians know who is in which group.
Randomisation ensures that the various patient groups in the trial are statistically similar, and blinding mitigates bias in how healthcare practitioners and participants in the trial behave4, as well as how outcomes are assessed or reported.
Observational data collected outside of the context of a clinical trial, by contrast, are often hopelessly confounded, which is why we see results like vitamin D supplementation failing to improve health outcomes for most patient groups across many randomised controlled trials, despite seemingly strong associations between vitamin D levels and morbidity or mortality in retrospective observational data. The value of randomised clinical trials is their ability to separate the true effect of a drug from chance effects, which include the regression to the mean that is often observed in episodic or cyclical conditions as patients are most likely to enrol in a trial when symptoms are near their zenith.
Trials are planned with a sample size that’s expected to be large enough to detect a statistically significant drug effect on the primary endpoint, if it exists. Typically, regulatory agencies require success (statistically significant improvement in a meaningful primary endpoint) in two independent phase III trials for approval. This requirement mitigates the risk of false positives due to chance or bias. Exceptions are sometimes made in situations when no good treatments are available on the market, or when the condition is so rare that there aren’t enough patients to run multiple trials. In these cases, drugs can either gain approval based on a single successful phase III, or even based solely on a phase II if the need is great enough and the early data sufficiently compelling.
Once the drug has shown its efficacy and safety in clinical trials, and regulators are convinced it can be manufactured consistently and safely at scale5, then the drug will be approved and given a marketing authorisation for a specific ‘indication’6 (indications describe the conditions and circumstances where the drug can be appropriately used).
Act 3: Pharmaceutical regulation from Janssen to today; regulation as a compromise between safety and efficacy
Drugs can, and frequently do, fail to advance at any stage in the development process, wiping out the investment up to that point, usually because they don’t work, but roughly a quarter of the time because of safety issues that weren’t picked up in preclinical testing. We cannot yet perfectly extrapolate human safety from preclinical models, like lab mice, which means humans must necessarily be exposed to some risk to move medical science forwards.
Biology is unpredictable, especially when the immune system is involved, and there is no guarantee that a drug that appears safe in animals will be safe in humans. The biotech firm TeGenero ran into this issue during first-in-human trials in 2006, when their antibody candidate TGN1412 overstimulated trial participants’ immune systems, leading to cytokine storms that left six previously healthy volunteers hospitalised with multiple organ failure, and one patient losing fingers and toes. This happened even though there were no signs of side effects in preclinical experiments, the dose administered was 500 times lower than the safe dose in animals, and there were no obvious deficiencies in the preclinical studies or manufacturing process.
A likely contributing factor to this dramatic difference in responses between animal experiments and human trial subjects is that the immune system develops throughout life and through exposure to a microbe-rich environment. An immunologically naïve lab mouse is not going to react in the same way a human would — not to mention that each human will have a different set of life experiences and immunological exposures. Subtle differences in biology between animal models and humans can lead to large differences in the toxicity of drugs: even though you and your dog are both mammals, chocolate is a treat for you and a toxin for them.
During preclinical testing we do the best we can to keep trial volunteers safe, although the risk can never be fully eliminated. There is a natural trade-off between the risks trial participants and broader society are willing to accept, and the regulatory burden placed on drugmakers to demonstrate safety. This extends to the post-marketing period as well. Clinical trials are usually too small to pick up rare side effects, like the risk of blood clots (3.2 reported per million doses) that ultimately sank the Johnson & Johnson COVID-19 vaccine7, and cannot practically be made big or long enough to catch them without making drug development uneconomical. The modern regulatory compromise was arrived at through a series of safety scandals, and each scandal has progressively tightened the regulatory ratchet. It is rarely loosened.
The idea that the pharmaceutical industry should be centrally regulated is a relatively new idea. Before the 1950s regulation was embryonic: it focused on preventing the sale of fraudulent, impure, or adulterated goods, or was limited to specific diseases or products. Before this regulation was introduced, many scam purveyors pushed literal snake oil, and other similarly useless, if not downright harmful drugs — though this was partly constrained by private efforts like the American Medical Association’s (AMA) drug evaluation program that ran from 1905 to 1955 in which association physicians reviewed and furnished drugs with ‘Seals of Acceptance’.
Norway and Sweden were some of the first countries to implement broad efficacy and safety legislation around the 1930s. In the USA, a pivotal moment came when over 100 deaths resulted from ingestion of an elixir of the widely used antibiotic sulfanilamide dissolved in poisonous diethylene glycol — a chemical similar to antifreeze. This led to the passing of the 1938 Food and Drug act in the US, which required that drug developers submit evidence to the FDA demonstrating the safety of their products before they could be marketed. The act also established sulfanilamide and other “dangerous” drugs as prescription-only which changed the patient-physician relationship and cemented the role of the physician as an intermediary agent evaluating the benefit-risk trade-off of drugs and prescribing them on behalf of the patient. Beforehand, patients would mostly buy drugs directly. A few countries, France included, also established their own panel of experts to review and approve new drugs prior to marketing authorisation around this time. Further safety scandals, including the deaths of nearly 300 people caused by tainted antibiotics, led the FDA to establish controls on manufacturing in 1941.
The rest of the world was slower to establish this sort of regulation, which changed following the thalidomide disaster. Thalidomide was introduced to most of the world as a treatment for anxiety, insomnia, and morning sickness without first being tested in pregnant women. The US was a notable exception, where FDA reviewer Frances Kelsey rejected the drug’s marketing application, citing a need for further safety studies. Tragically, thalidomide turned out to cause severe developmental defects in foetuses.
Once the safety issues became apparent, and before thalidomide could be removed from the market in 1961, over 10,000 babies would be born with truncated limbs and other birth defects, and thousands of extra miscarriages and abortions took place besides. The US responded in 1962 by amending the Food, Drug, and Cosmetics act to require that drug developers demonstrate efficacy in addition to safety — reasoning that every active drug carries risk, and therefore risk-benefit evaluations are critical. In similar fashion, the UK set up the committee on the safety of drugs in 1964 to evaluate efficacy and safety of new drugs, and formalised the regulatory process in 1968 with the Medicines act. Other countries followed, and by the 1980s, comparable regulation was widespread worldwide.
Since the 1960s, drug regulators have tried to draw a compromise between those who think regulations are too stringent, and those who think regulators are too lax and let unsafe drugs onto the market. Hearings in the 1970’s led by Senator Ted Kennedy accused FDA superiors of placing pressure on reviewers to approve drugs that were not sufficiently demonstrated as safe or effective. Taking the other position, economists such as Sam Peltzman argued that the 1962 amendments raised the cost to develop new drugs and put up barriers that harmed the public in excess of any harms avoided by delaying or rejecting new drugs. Haloperidol was one of the drugs affected by the FDA’s relative stringency compared to the rest of the world at the time: although haloperidol was used commercially in Europe from 1959 onwards, it was not approved in the US until 1967.
The challenges to the FDA’s harm avoidance mindset came to a head during the AIDS epidemic. Without effective treatments for AIDS, and facing certain death, activists stormed the FDA’s headquarters in 1988 carrying signs with slogans including “R.I.P. killed by the FDA”. Their demands included shorter drug approvals (as soon as after phase I), and the removal of placebo controlled trials, arguing that giving inactive placebos to patients with fatal diseases is unethical. The AIDS activists contended that experimental therapies are a form of health care, and as such they had a right to access them.

The urgency to act during the early days of the AIDS epidemic spurred the FDA to set up pathways to get AIDS drugs like azidothymidine (AZT) and didanosine (DDI) to market faster than they would otherwise. This laid the groundwork for the accelerated approval pathway, which was formally instituted in 1992.
The motivation behind accelerated approvals was that patients with terminal diseases like cancer and AIDS did not have the luxury of time to wait for new drugs to get to market, and should be able to elect to assume greater personal risk if they so wished.
The accelerated approval regulations allowed drugs to be approved based on a surrogate endpoint if they addressed a serious or life threatening condition with no good treatments available. The manufacturer could then confirm efficacy and safety later by conducting confirmatory post-marketing trials. Similar pathways were adopted some time later in other regions too, like the EMA’s conditional marketing authorization.
Surrogate endpoints are not a direct measure of the outcomes we really care about — how a patient ‘feels, functions, or survives’ — but they are measures meant to predict those outcomes down the line. The advantage of surrogates is that they are generally faster, easier, or more reliable to measure than standard endpoints. A cancer trial may assess changes in the size of the tumour as a surrogate, rather than the ultimately meaningful endpoint of patient survival; a cardiovascular trial might look at cholesterol levels or blood pressure. Blood pressure is an exemplary ‘validated’ surrogate endpoint: high blood pressure is highly predictive of strokes and other cardiovascular events. Measurable biomarkers (a portmanteau of ‘biological markers’) of disease are often repurposed as surrogates: I’ve already given the examples of blood pressure and cholesterol, another good example is the use of prostate-specific antigen (PSA) levels as a measure of response to prostate cancer treatment.
Not all surrogates are known to be valid predictors of disease outcomes, or they may only correlate weakly with eventual results on truly important outcomes. Regulators are justifiably sceptical of new surrogates, as encouraging results on surrogate endpoints in early trials often fail to pan out in larger confirmatory studies. Although, when used appropriately for diseases with few good options, approvals based on surrogates are a useful mechanism to get promising drugs to patients faster.
While the AIDS crisis and the subsequent sanctioning of surrogate-based approvals represented a rare relaxation of regulatory standards in response to external pressures, it seems to be broadly true that regulatory and societal tolerance for risk declines with time. We can see examples of this with grandfathered-in approvals for old drugs8. A common refrain is that paracetamol, with its marginal painkilling efficacy and risk of liver damage, would not be approved by regulators if it were discovered today.
In some sense regulators are in an impossible position, forced to mediate between groups with fundamentally incompatible worldviews; some consumers demand regulation, others demand access. Yet, the playing field is uneven: in the minds of the general public, the media, and policymakers, the ‘seen’ harms of unsafe drugs tend to overpower the ‘unseen’ harms of delaying or blocking a potentially beneficial drug’s path to market9. After all, dead patients cannot advocate for themselves.
History has shown that regulation is necessary to protect patients from unscrupulous firms taking advantage of them with unsafe or useless drugs, but there is a delicate balance to be found. Regulators are incentivised to be risk averse, and it’s hard to fight this general tendency, especially when the general public often supports exercising caution. Janssen himself noticed this issue, and was an advocate for a greater public acceptance of risk:
I think that [the general public can facilitate discovery by] having a more positive attitude towards drugs, looking more at a drug’s efficacy and less at the issue of risks. These days the emphasis seems to be almost exclusively on risk factors, even for necessary drugs such as the whooping cough vaccine in the United States. It is in deep trouble. Only one producer is left because the claim is made that there is one chance in a million to develop encephalitis in children. This is very strange because on the one hand everybody knows that we cannot do without this vaccine. On the other hand, this so called “risk” is scientifically not proven, it may only be a rumor. But a rumor is like an x-ray, it penetrates through almost everything…In one of the last issues of Science, there is a very interesting article about how risk is being perceived these days by the population. There is virtually no relationship with measurable risk. So that what I am talking about is the perception of risk, not real risk. This is a psychological phenomenon, I am sure. What should we do to speed up the process of discovery? We should educate the population in such a way that risk would be perceived as it is, and not in any artificial way. That can be and is very destructive.
While Janssen said this in 1986, little has changed since. The pharmaceutical industry is still viewed with broad scepticism by the general public, despite the good their drugs do. Vaccines in particular continue to fight against public perceptions of safety risks that are out of line with their actual objective risk.
Over the long-term, the trend has been to steadily impose tighter and tighter constraints on drugmakers. From where we stand today, we may only be a few more tightenings of the ratchet away from a world in which the barriers to entry for new pharmaceuticals are prohibitive. If the burden of satisfying the institutions that approve and pay for drugs were to exceed the rewards to innovation, investment in new drug discovery would be threatened.
Act 4: The cost of development is catching up with us, or, why are drugs so expensive to make now?
The biopharmaceutical industry expends huge sums shepherding drug candidates through the development gauntlet and satisfying regulatory requirements. In 2022, the industry spent around $200 billion on R&D, more than four times the US National Institute of Health’s (NIH) budget of $48 billion. Pharmaceuticals is the third most R&D intensive sector in the OECD countries.
The bulk of that spending goes towards clinical trials and associated manufacturing costs; roughly 50% of total large pharma R&D spend is apportioned to phase I, II, and III trials compared to 15% for preclinical work. While early phases may cull more candidate compounds in aggregate, the cost of failure is highest during clinical development: a late-stage flop in a phase III trial hurts far more than an unsuccessful preclinical mouse study. By the time a drug gets into phase III, the work required to bring it to that point may have consumed half a decade, or longer, and tens if not hundreds of millions of dollars

Clinical trials are expensive because they are complex, bureaucratic, and reliant on highly skilled labour. Trials now cost as much as $100,000 per patient to run, and sometimes up to $300,000 or even $500,000 per patient for resource-intensive designs, trials using expensive standard of care medicines as controls or as part of a combination, or in conditions with hard-to-find patients (e.g., rare diseases). When these costs are added on top of other research and development expenditures, like manufacturing, a typical phase I program with 20-80 trial participants can be expected to burn around $30m. Phase III programs, involving hundreds of patients, often require outlays of hundreds of millions of dollars. Clinical trials in conditions where large trials with tens of thousands of patients are standard, such as cardiovascular disease or diabetes, can cost as much as $1 billion.
| Brand name | Drug type | Disease (first approval) | FDA approval year | Phase I R&D spend | Phase II R&D spend | Phase III R&D spend | Total R&D spend (Phase I-III) |
|---|---|---|---|---|---|---|---|
| Belviq | Small molecule | Obesity | 2012 | $75m | $136m | $607m | $817m |
| Nuplazid | Small molecule | Parkinson’s disease psychosis | 2016 | $35m | $107m | $373m | $515m |
| Vimizim | Biologic (enzyme replacement) | Rare congenital enzyme disorder | 2014 | $33m | $114m | $346m | $493m |
| Galafold | Small molecule | Rare congenital enzyme disorder | 2018 | $28m | $44m | $324m | $396m |
| Xerava | Small molecule | Abdominal infections | 2018 | $31m | $32m | $325m | $388m |
| Ultomiris | Biologic (monoclonal antibody) | Rare blood disorder | 2018 | $9m | $71m | $275m | $354m |
| Alunbrig | Small molecule | Lung cancer | 2017 | $10m | $243m | $98m | $351m |
| Ocaliva | Small molecule | Rare liver disease | 2016 | $28m | $24m | $299m | $351m |
| Trulance | Peptide | Constipation | 2017 | $7m | $88m | $253m | $348m |
| Aristada | Small molecule | Schizophrenia | 2015 | - | $47m | $282m | $329m |
| Ingrezza | Small molecule | Movement disorder | 2017 | $14m | $108m | $202m | $323m |
| Veltassa | Small molecule | Elevated blood potassium | 2015 | $61m | $68m | $191m | $319m |
| Gattex | Peptide | Short bowel syndrome | 2012 | $2m | $55m | $253m | $309m |
| Tymlos | Peptide | Osteoporosis | 2017 | $28m | $21m | $256m | $305m |
| Iclusig | Small molecule | Rare blood cancer | 2016 | $50m | $223m | Approved off phase II | $272m |
| Brineura | Biologic (enzyme replacement) | Batten disease (rare genetic disorder) | 2017 | $42m | $220m | Approved off phase II | $263m |
| Eucrisa | Small molecule | Eczema | 2016 | $30m | $49m | $129m | $208m |
| Elzonris | Biologic (fusion protein) | Rare blood cancer | 2018 | - | $168m | Approved off phase II | $168m |
| Viibryd | Small molecule | Depression | 2011 | - | $7m | $146m | $153m |
| Mepsevii | Biologic (enzyme replacement) | Rare congenital enzyme disorder | 2017 | $13m | $13m | $121m | $146m |
| Rhopressa | Small molecule | Glaucoma | 2017 | $14m | $25m | $98m | $136m |
| Vitrakvi | Small molecule | Rare cancer subtype | 2018 | $15m | $104m | Approved off phase II | $119m |
| Folotyn | Small molecule | Lymphoma subtype | 2009 | $19m | $89m | Approved off phase II | $108m |
| AVERAGE | $27m | $89m | $254m | $312m |
Because executing late stage clinical trials and manufacturing enough of the drug to cover them is so expensive, companies prefer to manage risk by conducting studies sequentially, even though many steps could in principle be done in parallel.
A major reason that COVID-19 vaccine development was so fast was not because shortcuts were taken, but that the funding from operation warp speed and advance purchase agreements allowed companies to parallelize much of the process, scale up manufacturing early, and jump quickly into phase IIIs because they were insulated from the financial risk of failure. Early trial phases were combined in multiphase designs, Pfizer commandeered existing manufacturing infrastructure and repurposed it for COVID-19 vaccine production, and employees and regulators worked around the clock. The FDA and other regulators took reviewers off of non-COVID-19 drugs and redeployed them to review the COVID-19 vaccines; there were essentially no delays in safety reviews that you would otherwise see in other clinical trials. The little delays that crop up in development were powered through with extra manpower and resources: at one point during Operation Warp Speed the military recovered a vital piece of equipment needed to manufacture Moderna’s vaccine from a stalled train, and put it on an aeroplane so it could arrive in time. While the vaccines were approved under expedited emergency use regulatory pathways, they were nevertheless rigorously tested. Allocating such extensive resources for every new drug, as was done during the pandemic, is unsustainable and comes with substantial opportunity cost.
In business-as-usual times, the industry’s expenditure on drug R&D nets us about 40 new US FDA drug approvals a year — and a similar number (though not always exactly the same drugs) approved by the equivalent agencies in other regions, as well as some new indications for existing drugs.
All that money spent by the industry on R&D appeared to go a lot further in the past10. Despite continued growth in biopharmaceutical R&D expenditure, we have not seen a proportionate growth in output. Industry R&D efficiency — crudely measured as the number of FDA approved drugs per billion dollars of real R&D spend — has (until recently) been on a long-term declining trajectory11. This trend has been sardonically named “Eroom’s law” — an inversion of Moore’s law. Accounting for the cost of failures and inflation, the industry now spends about $2.5 billion per approved drug, compared to $40 million (in today’s dollars) when Janssen was starting out in 1953.
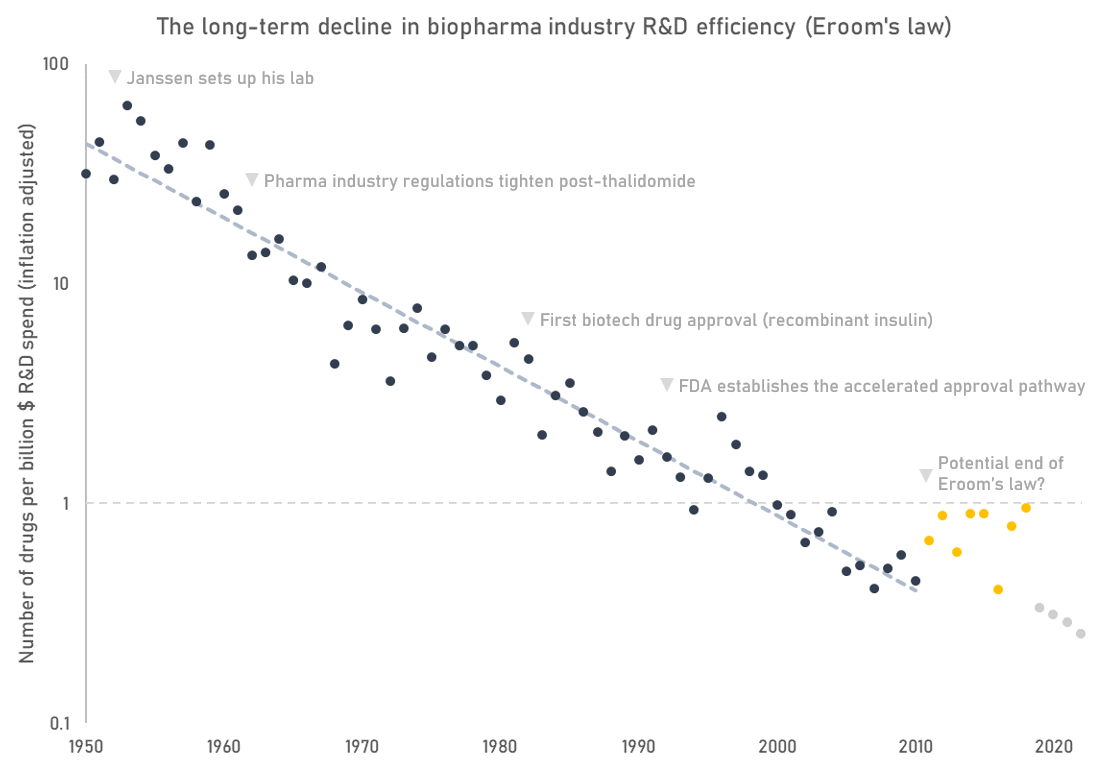
The number of drug approvals is just an indicator metric. It only gives a hint of how we are doing on the actually important question: how much improvement in quality and length of life are we getting for our money? We don’t have the data to answer that definitively, but proxy metrics suggest the industry’s impact is waning. The aggregate profitability and return on investment of the pharmaceutical and biotech sector has declined since the 1980s, suggesting that the industry may be delivering less value than in the past. Estimated industry return on investment is close to turning negative. Development stage programs today focus disproportionately on less prevalent diseases where pricing power can be maintained, and not the areas with the highest global burden of disease. Fewer breakthroughs, and more incremental advances.
Ultimately, the decline in R&D productivity affects us all because we’re getting fewer drugs than we might otherwise. Drugs are one of the most cost-effective healthcare interventions, and have significantly raised lifespan and healthspan. In the 100 or so years in which the pharmaceutical industry has existed, we have seen millions of deaths from communicable diseases averted due to vaccinations and antibiotics, and large declines in the age-standardised cancer and cardiovascular death rates12. Increasingly, new treatments are transforming the lives of patients with rare and devastating genetic diseases such as cystic fibrosis and spinal muscular atrophy.
Yet there is much still to do. Cardiovascular disease, cancer, and mental health remain leading causes of death and disability, age-related neurodegenerative diseases like Alzheimer’s are growing in prevalence in step with demographic shifts, and only a tiny fraction of the estimated 10,000-plus rare diseases have effective treatment options.
Even though we’re spending more money than ever before, historical statistics on drug candidate failure rates suggest that we haven’t really gotten much better at developing drugs that succeed where it counts — in clinical trials. The real bottleneck is not finding drug candidates that bind and modulate targets of interest, it’s finding ones that actually benefit patients. Almost paradoxically, despite huge improvements in the technologies of drug discovery, the rate of new drug launches has hardly shifted in 50 years. High-throughput screening, new model systems, machine learning, and other fancy modern techniques have done little to change the statistic that 9 in 10 drug candidates that start clinical trials will fail to secure approval.
What’s behind this ‘Red Queen’ effect, where we seem to be expending more and more resources to keep running at roughly the same speed?
For one, as we’ve seen across scientific fields, new ideas are getting harder to find. There are more academic researchers than ever — 80,000 in the 1930’s vs. 1.5 million today in the USA — yet we have not seen a proportionate growth in the rate of meaningful discoveries. This may be because ideas are getting inherently more difficult to find, or it may be that the institutions and processes of science have become less effective: bogged down in bureaucracy, sclerotic, chasing the wrong metrics, and thereby limiting the impact of individual researchers.
The biopharmaceutical business is built on top of basic discoveries, and so it is not immune from this general trend afflicting all of science. Without the discovery of methods to stabilise coronavirus spike proteins and enhance immunogenicity prior to the pandemic, it’s unlikely that we would have had effective vaccines for COVID-19 as fast as we did. Imatinib, a breakthrough targeted therapy for a rare blood cancer, was predicated on the discovery of the mutant protein produced by the “Philadelphia chromosome” rearrangement. In support of the ‘low hanging fruit’ argument is data showing changes in the landscape of drug targets over time: compared to past decades, drugs in development are now much more frequently going after targets that would have historically been viewed as intractable. Modern protein targets are more likely to be disordered, with shallow or non-existing pockets for small molecules to bind, or otherwise difficult to interact with.
The more important reason for the decline in R&D efficiency, however, is that it is not enough for drugs to simply be novel and safe, they must also improve meaningfully over the available standard of care, which may include a large armamentarium of effective and cheap older drugs.
This is the so-called ‘better than Beatles’ problem. Imagine if in order to release new music it needed to be adjudicated as better than ‘Hey Jude’, or ‘Here comes the sun’ in a controlled experiment. New experimental music might have a hard time getting past the panel, and wouldn’t have the chance to refine its sound in future iterations. The situation for new drugs is somewhat analogous.
There are counterexamples — antibiotics, for example, where greater use steadily creates resistance and depreciates the value of the original idea — but by and large, the jump from no drug to first drug is always larger than the jump from first drug to second drug. This means that in some sense a decline in industry R&D efficiency is not such a bad thing: it reflects in part the fact that we are such a rich society that we can afford to invest increasingly large sums to make increasingly small improvements in health.
Along with drugs getting harder to find, it is also getting more expensive to run the clinical trials required to test them. Today’s astronomical trial costs are a consequence of the trend towards trials being larger, more complicated, enrolling more narrowly defined patients across more sites, and more resource intensive than they used to be.
Across many conditions, trials are getting bigger. The need to demonstrate benefits over the standard of care means effect sizes shrink over time, which means larger trials are required to have sufficient statistical power to differentiate the new treatment from the old one. Current lipid-lowering treatments for cardiovascular disease, primarily statins, are so effective that the improvements achieved by the PCSK9 inhibitors — a new class of cholesterol-lowering drugs — on top of statins required a massive development program to demonstrate. The ODYSSEY trial that showed alirocumab treatment led to a 15% relative risk reduction in mortality in combination with statin therapy enrolled just under 19,000 patients at a high risk of additional cardiovascular events and took around 5 years to run at a cost of around $1 billion.
Increasing sample sizes across the board means the limited supply of trial volunteers imposes a severe constraint on the feasibility of clinical research. Only 6% of cancer patients take part in clinical trials nationally in the US, for instance, and the number is generally lower in other countries and for other conditions. To get enough patients to fill up large trials companies need to conduct trials at multiple sites. The more sites involved in a trial, the greater the logistical complexities involved in coordinating that the protocol is executed appropriately across sites, the data is collected to a good standard, and the drug is distributed to all sites as needed. This all increases costs. More sites also increases variance in execution, and improper trial conduct can delay or even sink a development program. According to data from Tufts university, >80% of trials fail to recruit on time, actual enrolment times are typically around double the planned timelines, and ~50% of terminated trials result from recruitment failures. An estimated 11% of trial sites fail to recruit a single patient, and another 37% don’t reach their target enrollment criteria.
Many patients are willing to take part in clinical trials in principle, but awareness is poor. About 50% of the time when patients are invited to clinical trials they accept, but 90% are never invited to participate, mainly because most patients are not treated in settings that conduct trials. Patients are also not necessarily aware of or educated about the benefits of trials, and how they may enable them to access a high standard of care. Leading clinical research centres often have too many studies and not enough patients. When it comes to the trial itself, the site may be far from where the patient lives, requiring them to travel or even relocate for the duration of the trial — without adequate support for doing so.
Simply opening new trial sites is not an easy solution, because few sites have the capabilities to handle complex trial protocols or the staff trained in the good clinical practice (GCP) processes required to execute trials to standard. Trials increasingly require sites to carry out complex and resource intensive procedures and tests on expensive equipment, which may not be available outside of the best-resourced centres. Regulators are also often reluctant to accept data from sites and regions where they are unsure of the quality of the data.
Even if new sites could be spun up easily, there’s no guarantee they would have the patients, especially rare disease patients who tend to be sparsely distributed outside of specialised centres. Remote trials — collecting data at-home with smartphones and other digital tools — are one oft-touted solution to increase access to trials. However, there are many procedures that cannot be done remotely, like blood draws. Unreliable data and high dropout rates are further problems with remote trials.
The difficulty of recruitment is exacerbated by significant duplication of effort in the industry, with many similar drugs chasing the same targets and competing for the same patients. This is a problem because it can lead to a limited pool of patients being split up between trials meaning that no single trial recruits enough patients to properly test a hypothesis, increasing timelines for all.
When drug developers can’t secure their target level of funding or patients for their trial they will often go ahead anyway, even though underfunded and under-enrolled trials lead to wasteful research. An analysis of 2,895 COVID-19 therapeutic trials by Janet Woodcock of the FDA concluded that only 5% were randomised and adequately powered, implying 95% of the effort was unlikely to yield actionable information. Wasteful spending is a feature of the industry at large; take for example the estimated $1.6 billion spent on trials for 16 different IGF-1R inhibitors in cancer, none of which were approved. Biotech executives are incentivized to run trials of their compounds even if they can’t secure the funds to sufficiently enrol them, or if someone else is already testing a similar drug, as the alternative is giving up their job. Kicking the can down the road can be personally lucrative, and an unclear result can be an opportunity for further delays. Every patient who volunteers for a trial is a precious resource, and duplicative and underpowered research is a regrettable waste of their generosity.
Further exacerbating the issues is the increased bureaucratic complexity involved in setting up trial sites today, as compared to Janssen’s time. Site identification and setup is a complex lengthy process that regularly takes as long as 8 months and requires extensive documentation and ethics board review. One frequent source of complaints, for instance, is the process of obtaining informed consent. The process is burdensome for patients and physicians, and requires each participant to read through multiple lengthy forms that they frequently do not understand. Informed consent documents are bloated not in service to patients, but in an attempt to address unclear regulations and avoid potential litigation.
When Janssen was conducting trials, bureaucracy was minimal. Ethical and enrollment decisions were primarily made based on individual physician discretion. Initial tests were set up quickly; Janssen could drive over to a colleague and administer a drug to a patient in a few days. The first trials of haloperidol were run rapidly in a psychiatric ward in Liege:
The next step [after testing haloperidol in animals] was to test the effect on a human being” Janssen said. “I was lucky enough to meet the very capable Dr Bobon in Liege who introduced me to a 16 year old boy, recently admitted to his clinic and showing all the symptoms of paranoid schizophrenia. Dr Bobon agreed to give him haloperidol and he calmed down; the effect was amazing.
Shortly after these encouraging initial tests in Belgium, haloperidol was distributed to psychiatrists in seven European countries, with relatively few stipulations about how they should use it:
In those days double-blind studies were not yet designed. I gave the drug independently to eight well-known psychiatrists, in seven countries — Sweden, Denmark, Belgium, France, Germany, Portugal, and Spain — and provided them with the pharmacological and toxicological data. I explained my expectations about the therapeutic effectiveness and gave them an idea of what I thought was the optimum dose.
About a year later, all investigators had got the same clinical impression, namely the control of hallucinations and motoric control. They came out with the same dose recommendations. The results of many double-blind tests later on were mainly a confirmation of the first impression of these psychiatrists
Favourable results from initial trials were rapidly disseminated to physicians through journal publications, and swift application in actual clinical practice followed.
Of course, the limited oversight in early trials came with its own set of problems. Patients were entered into trials without proper consent and were exposed to excessive risk. Serious safety events were relatively common, and would be unlikely to be acceptable today: such as an early chloramphenicol trial in which 45 premature babies died, that was cited by Ted Kennedy as an example of life-threatening medical research. Trials in the mid-1900s were also often conducted using underprivileged individuals who could not provide proper consent — the Tuskegee syphilis study, or the many studies of vaccines conducted in people with severe mental disabilities being notable examples. I don’t want to suggest we should go back to this level of laissez faire research, but there is a trade-off to recognise as the looser paradigm of trial regulation and liberal testing in human subjects is likely to have meaningfully accelerated research.
The complexity, bureaucracy, and sheer amount of money involved in running clinical trials today has created an industry of its own. A second order consequence of the demand exceeding the supply for skilled and trusted operators and sites with patients is that the outsourced contract research organisations (CROs) and clinical trial sites who execute trials on behalf of biopharmaceutical companies can extract extra rent with little downside, and aren’t forced to innovate to improve processes. Because trials are so expensive in money and time that there is a natural conservatism when it comes to adopting new processes and tools. Drug developers are generally reluctant to take on regulatory risks when established precedents in trial design and execution exist and so are likely to rely on tried and tested processes and vendors. The barriers to entry and risk aversion are so high that they can block upstarts from coming in with new methods and getting sufficient funding and scale to positively disrupt the industry.
It’s not just trial operations which consume inordinate amounts of time and money. Manufacturing for drugs entering human trials are controlled by strict good manufacturing practice (GMP) regulations, which requires developers implement strict quality controls on consistency, potency, impurities, and stability before their drug can be tested in humans. These regulations are important for safety and for making sure that the drug that is tested is the same as the one that is eventually approved. Still, maintaining GMP compliance for early clinical trials is nevertheless a substantial drain on the limited resources of fledgling biotechs.
All these factors — stiffer competition, regulation, bigger and more complex trials, expensive manufacturing — conspire to erect barriers to entry that are far more expensive for drugmakers to overcome than in the past. When Janssen was asked in a 1986 interview if he could have developed haloperidol in the 1980s as easily as in the 1950s, he said:
No. Certainly not. Because the bureaucratic requirements would make it impossible both in terms of time and in terms of money. When I began, bureaucratic requirements were minimal as compared to what they are now. Today we have to do chronic toxicity work, we have to explore the metabolism. We have to know what a mechanism’s action is before we can even start clinical testing. In those days, it was simply a matter of our own conscience. There was no bureaucrat looking at our fingers. Today we have to fulfill all these bureaucratic requirements. Whether we like them or not. This is a dictatorial system we have to submit to. It’s extraordinarily expensive. And young people no longer have the chance that I had in 1953 to start a new company. It’s out of the question. Not even with several thousands of times the capital I had.
Yet, even though there are major forces pushing against drug developers, there is a sense that the industry is still underperforming, and that it could do more. One reason for optimism can be seen in the recent flattening of the slope of Eroom’s law following decades of declining productivity. It remains to be seen whether the recent uptick is a sustained turnaround or not. The pessimistic view is that it is illusory, a result of how drugmakers have side-stepped fundamental productivity issues by focusing on developing drugs for niche subpopulations with few or no options where regulators are willing to accept less evidence, it’s easier to improve on the standard of care, and payers have less power to push back on higher prices: rare disease and oncology in particular. It’s no coincidence that investment has flowed into areas where regulatory restrictions have been relaxed and accelerated approvals are commonplace: 27% of FDA drug approvals in 2022 were for oncology, the largest therapeutic area category, and 57% were for rare/orphan diseases.
There is however, a more charitable and optimistic take for the flattening and possible reversal of Eroom’s law. The first possibility is that advances in basic science are finally being widely adopted in the drug development process and bearing fruit. Historically, it takes upwards of 20 years for new drug targets to lead to new medicines. Consider that the sequencing of the human genome was completed in 2003; genomics research has by now improved our understanding of many relatively simple monogenic genetic disease, and has identified new targets for more common conditions through genome-wide association studies (GWAS) that look for associations between gene variants and disease phenotypes in large populations. The PCSK9 inhibitors alirocumab and evolucumab, as an archetypal example, were developed after screening for genetic mutations in families with elevated cholesterol levels identified the PCSK9 gene as a key driver of cholesterol regulation. Drug programs with genetic support are more likely to succeed, and we may have only recently truly started to benefit from our improved understanding of human genetics.
The rise of biologics
Another possible driving force behind recent productivity gains is the rise of biologic drugs. Traditional pharmaceuticals of the kind produced by Janssen are known as ‘small molecules’. The size cut-off that defines a small molecule is loose, but a good rule of thumb is small molecules are chemically synthesised and are small enough to be reasonably visualised by a chemical skeleton diagram. Biologics have a mass hundreds or thousands of times larger than small molecules and include proteins, cells, or other macromolecules like mRNA produced in living organisms. Unlike small molecules, biologics are grown in living organisms, not chemically synthesised.
The recombinant DNA revolution of the 1980’s led by Genentech and its peers enabled biologic drugs like proteins and peptide hormones to be produced at scale by inserting the genes for human proteins into bacteria and growing them in huge bioreactors.

The development of recombinant ‘humanised’ insulin was a great early triumph of the nascent biotech industry. Before the recombinant version was approved in 1982, insulin had to be extracted from the pancreases of pigs and cows, and injecting it frequently caused allergic reactions.
Since the 80s, the biotech industry has grown in relative importance compared to the traditional medicinal chemistry of small molecules. Biologic drugs are increasing as a share of FDA approvals, and as of 2022, are close to overtaking ‘traditional’ small molecules. As medicinal chemistry came to displace natural product discovery, so is biotech coming to displace small molecule medicinal chemistry.
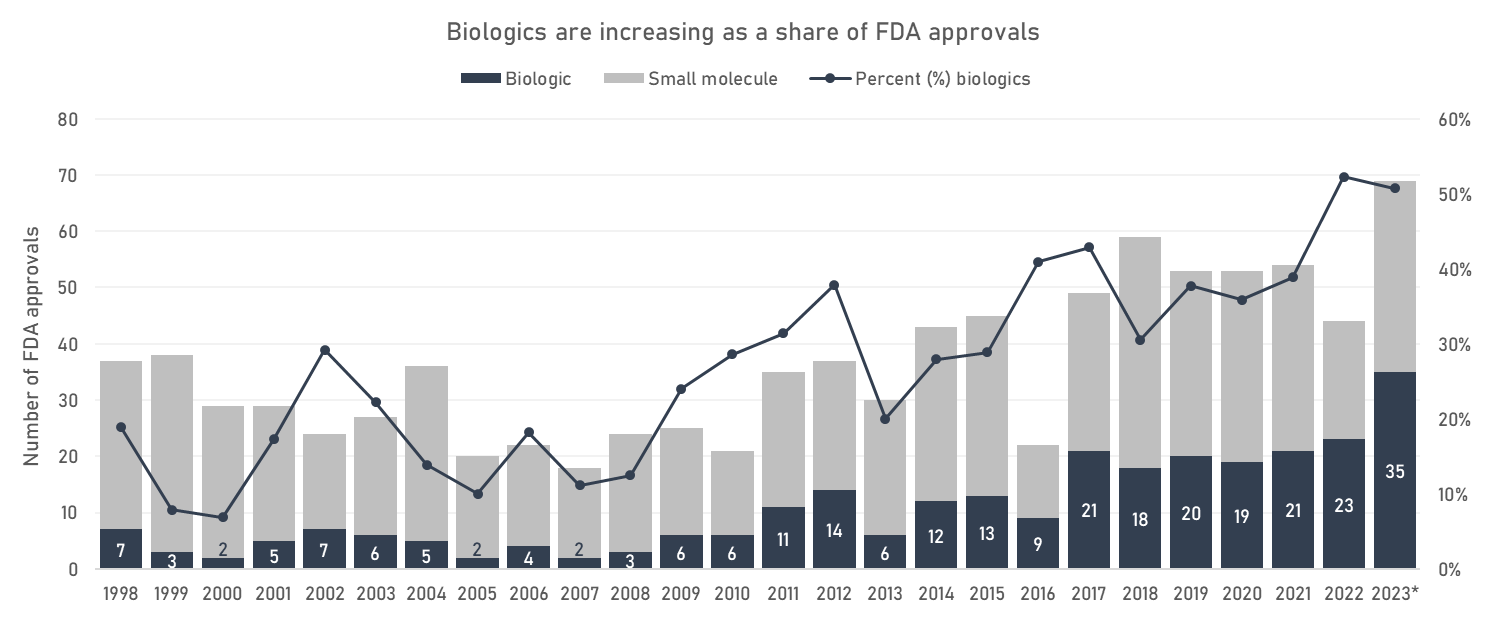
The most therapeutically and economically important class of biologics so far has been the monoclonal antibodies. Monoclonal antibodies are similar to the antibodies your body produces in response to an infection, but intentionally raised against disease-relevant targets and produced in immortalised cell cultures. The first therapeutic monoclonal antibody, an immunosuppressant for transplant patients, was FDA-approved in 1986. It took another decade or so for the technology to mature and to work out the manufacturing kinks; the second monoclonal antibody was not approved until 1996. Now they are an established class of drugs in oncology, immunology, and other disease areas: the FDA approved its 100th antibody in 2021. Of the top 10 revenue drugs in 2022, 3 were monoclonal antibodies.
RNA therapeutics are now coming into their own as well. Small interfering RNA — a technology capable of silencing production of specific proteins for long periods of time — is now finding multiple clinical applications. Inclisiran, a single injection that robustly knocks down PCSK9 and lowers cholesterol, only requires a single injection every 6 months compared to every 2 weeks for alirocumab. The adaptability of mRNA allowed the COVID-19 vaccines to be designed in two days on a computer after the virus sequence was made public. It’s easy to forget that prior to the pandemic it wasn’t clear that mRNA technology would work at all.
Even though some classes of biologics are now established, we are still in the early days of products the EMA refers to as advanced therapy medicinal products (ATMPs): gene & cell therapies, gene editing, or replacement tissues. These treatments have the potential to durably cure diseases, and early success stories are encouraging. Gene therapies have provided near-cures to patients with genetic disorders like haemophilia, sickle cell disease, and spinal muscular atrophy. Immune cells have been extracted, genetically edited outside the body, and reinfused, leading to durable cures of hard to treat childhood cancers. The first CRISPR therapy was approved by the FDA towards the end of 2023.
Unlike small molecules, ATMPs are immature technologies: they are more complicated and expensive to manufacture to standard, the best means of applying them is not yet fully understood, and the industry has not yet had time to optimise the manufacturing processes and develop the institutional knowledge that exists after fifty-plus years of small molecule development. ATMPs also tend to be exquisitely personalised and often even manufactured bespoke for each patient, which means smaller markets and greater process investment. Scaling ATMP delivery is consequently much more complex, resource-intensive, and procedure-like compared to a traditional pill in a bottle. For an extreme example take Bristol Myers Squibb, which has treated about as many patients with its cancer cell therapies as it has employees working on manufacturing and delivering said treatments (~4000 each).
Biologics today mostly repurpose genes and proteins that already exist in nature and add them back in a therapeutic context. Looking towards the horizon, the bioengineering toolbox continues to expand. We are starting to design our own biologics de novo, rather than relying on what we can mine from genomes or immune systems. Just as Janssen was a pioneer of the industry’s shift away from natural products, the next Janssen may be someone who pioneers the rational design and engineering of biologics that don’t exist in nature.
Act 5: Reinvigorating biomedical productivity
If we adopt the optimistic take on the levelling off of Eroom’s law — and with it the belief that biomedical stagnation is not inevitable — what can we do to encourage the reversal to continue?
Ultimately, we have three ways to get the most out of our incremental future research dollars:
- We can raise the success rate of clinical trials by improving the quality of input molecules
- We can make it cheaper to develop drugs by relaxing certain regulatory evidence or manufacturing standards, or making clinical trials more efficient
- We can incentivise types of research and development activity that have high potential to pay off in the future, such as gene therapies, even if development is currently relatively uneconomical
Improving the quality of clinical trial inputs
The most significant factor preventing us from taking better molecules into clinical trials is not our ability to design arbitrary molecules, but our (lack of) understanding of underlying disease biology. In other words, we often know how to make something, but not what to make. Hence, the most important advancements in drug candidate selection are likely to come from improvements in how we collect, analyse, and use biological data.
Expanding investment in improved disease models that are better able to predict how a drug will perform in humans is one promising avenue. Along these lines, Roche announced a new research institute focused on human organoids in May of this year, which aims to use ‘mini-organs’ derived from human cells to better recapitulate human biology. Organs-on-chips, perfused isolated whole organs, or patient-derived tissues are other examples of the growing menagerie of model systems with the potential to meaningfully improve preclinical-to-clinical translation success rates.
My personal bias is that the industry should recognise the limitations of the reductionist target-based screening approach and do more phenotypic screens with a wider diversity of input molecules (including obscure natural products). Most approved small molecule drugs were actually discovered by phenotypic screens — like the one that Janssen used to find haloperidol. Better models that are easier to work with could help industrialise this sort of phenotypic screening.
There are also potential benefits to be had from the increasing recognition of the importance of high-quality decision processes around advancing drug candidates through development. For example, AstraZeneca’s 5R’s framework. The 5R framework emphasises a set of criteria associated with program success, such as selecting targets with predictive biomarkers, and identifying the most responsive patient populations. While perhaps not as exciting as technological solutions, improvements in industry decision making processes could go a long way towards improving efficiency. A key way this is likely to manifest is through improvements in how we take advantage of the explosion in biological data collection. The value that genomic validation confers to a drug program is now widely known, and we have likely only scratched the surface of targets which we can mind from expansive datasets like those in the UK biobank.
Recent advances in artificial intelligence will hopefully eventually trickle into biological applications. Although many in the industry are sceptical, and might reasonably counter that useful AI tools have already been adopted by drug discovery teams for years — just without the AI branding. While there have been successes with protein structure prediction, notably AlphaFold, and some success with molecular design e.g. small molecules and proteins, it will take time for deep learning systems and training data to mature enough to be useful for the truly value-adding task of predicting safety and efficacy in humans.
Even though there is plenty of space to improve quality of clinical trial inputs, developing better drugs is going to remain an inherently challenging scientific problem. Near-term productivity gains may be more likely to come from policy adjustments that make it cheaper and faster to develop drugs. Especially in the context of emerging technologies or diseases with a high unmet need.
Making drug development cheaper
A promising approach to modernise regulatory evidence requirements in a way that benefits patients with few options is to adopt a regulatory framework that relaxes approval standards in a principled, consistent way in accordance with unmet need. Andrew Lo of MIT has suggested a Bayesian threshold for clinical trial result significance, which adapts the standard of evidence required to incorporate the need for new treatments and the risk of exposing patients to ineffective, or harmful, drugs. His analysis suggests the standard threshold for statistical significance (p≤0.05) is plausibly too stringent for lethal diseases with few good options like pancreatic cancer, but may actually be too relaxed for conditions with good treatment options like diabetes. Regulators could implement this framework by publishing guidance on varying significance thresholds by indication, which would provide more predictability than the current case-by-case approach. Lower thresholds could also help alleviate trial recruitment issues in rare or high-unmet need diseases by allowing for smaller trials.
Another option to reduce development costs is to expand the use of accelerated approvals and surrogates, and to increase investment in the discovery and validation of novel surrogates. Surrogates and accelerated approval have been used most heavily in cancers and rare diseases, and while we have seen marginal or questionably useful drugs approved — 50% of cancer drugs FDA approved on the basis of a surrogate outcome through 2008 and 2012 failed to show a survival benefit in confirmatory studies — these are also areas that have seen substantial gains in patient outcomes since the pathway was instituted. Mortality rates in breast cancer, for instance, where multiple important drugs like Xeloda, Femara, Ibrance, and Enhertu received accelerated approval, have decreased to such an extent that metastatic breast cancer is now considered by some to be a manageable chronic disease. While it wouldn’t be accurate to attribute these gains solely to accelerated approvals (screening and lifestyle changes also play an important role), it is fair to say that the accelerated approval pathway brought important drugs to patients years faster than they might otherwise have been.
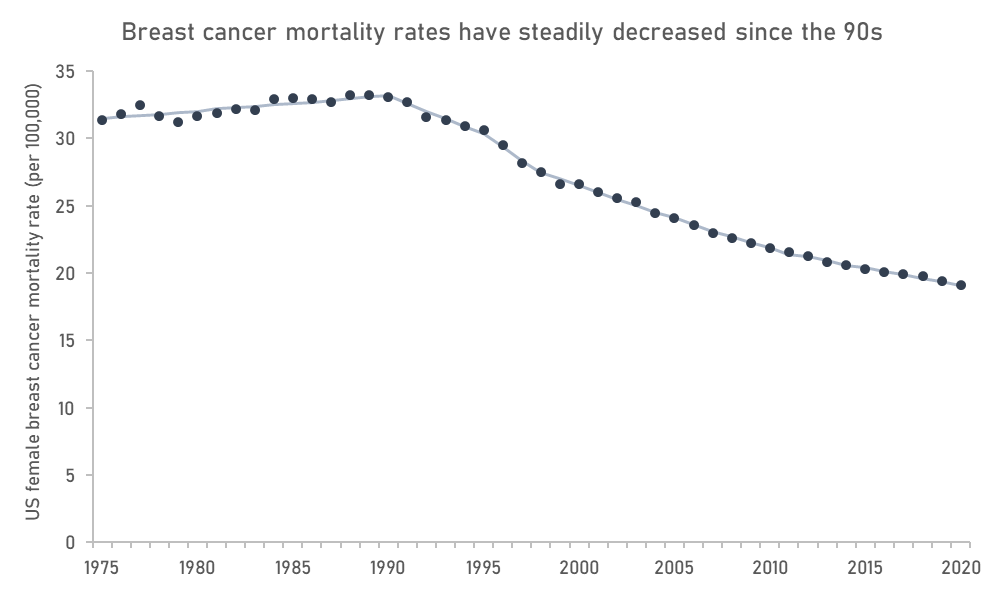
New surrogates also have the potential to unlock investment into conditions which otherwise require lengthy trials to test. Because patents have fixed term lengths, with small extensions to compensate for the year or so when the drug is under regulatory review, drug developers are incentivized to go after conditions where clinical trials can be completed quickly, all else being equal. When certain disease mechanisms take decades to play out it can be economically infeasible to develop drugs for these conditions without the availability of a validated surrogate. This is the biggest issue with developing drugs targeting ageing at the moment. It’s not that we lack promising ideas for interventions, they will just take so long to test, at such a huge cost, that the potential return on investment is limited. AI may have a concrete application here by mining voluminous data for new biomarkers that can be used as surrogates of slow-progressing diseases, cutting development time from decades down to years.
Greater reliance on surrogates necessarily transitions data collection from clinical trials to real-world settings, and this approach presents its own challenges. Data collected in the real-world setting is less robust than that collected in a clinical trial, so cannot fully compensate for true confirmatory randomised controlled trials. However, companies have previously taken advantage of the accelerated approval pathway by slow-walking the completion of confirmatory clinical trials after securing approval.
The adoption of more relaxed approval standards should therefore be balanced with stricter enforcement of drug removal when their benefits fail to be confirmed in a timely fashion. FDA reforms in 2022 are a step towards this, empowering the agency to require confirmatory studies be in place before accelerated approval is granted, and to expedite withdrawal if insufficient progress is made in confirmatory studies. Rebate or outcomes-based payment agreements could also be put in place if the drug’s efficacy does not hold up in the real world, in order to reduce incentives to seek accelerated approval of high-priced yet ineffective drugs. Improvements in data collection infrastructure and better data integration, including registries and electronic health records, will also be important to track the performance of drugs in the real world.
If regulators are to be tasked with greater post-marketing surveillance, they will also need more resources. The FDA and other regulatory bodies are understaffed and underfunded relative to the volume of applications, leading to delays, especially when it comes to people with experience in cell and gene therapies. Increasing funding for regulatory bodies would help them attract and retain staff, and respond to sponsors in a more timely fashion. Increases in funding, however, should be accompanied by greater accountability for delays in review timelines.
As a more general point, it would help if regulators could be more predictable and transparent in their decision making. In a survey of drug and device industry professionals, 68% said that the FDA’s unpredictability discouraged the development of new products. It can be hard to predict how regulators will react to a certain dataset in the context of high unmet need, so companies can be inclined to ‘submit and pray’, even after receiving negative feedback on the data package from regulators during prior interactions.
Easier access and more frequent interaction with regulators and payers prior to approval, and more explicit guidance on what is and is not required as part of a clinical trial design would help companies minimise the inclusion of unnecessary screening criteria, tests, and endpoints, thereby reducing costs and timelines. In an ideal world, it would not take 60 days from the time of request to meet with the FDA or EMA for advice on a development program. Limited transparency on expectations means companies today can be inclined to over-engineer the design of trials in case regulators or payers request certain datasets in future.
A straightforward start to improve transparency across the industry would be for the FDA to disclose the formal ‘complete response letters’ (CRLs) issued when they reject a drug which contain the reasons for rejection. Making this information public would give future developers insight into the regulator’s thinking on a disease, with minimal downsides. How companies represent their CRLs to the broader market today is often misrepresentative of the actual reasons for rejection, potentially misleading patients as well as future investors and drug developers in the indication.
Manufacturing requirements for complex products may also benefit from some relaxation of standards early in development. Small molecules are relatively simple to manufacture, but cell and gene therapies are complex and there are few people available with necessary expertise. Disruptions in development due to manufacturing issues are common for cell and gene therapies relative to more mature drug classes, often delaying programs for a year or more as companies collect further analytical data.
Contributing to these issues are the dozens of analytical assays that gene or cell therapy companies are forced to run to assess quality and release a batch, even during early clinical trials. In seeking to address bad actors, regulators can also often inadvertently punish the rest of the field. Imposing blanket, rather than targeted, rulings is often damaging. This has happened in cell and gene therapy, where regulators have unilaterally required additional quality assays for release to address aspects of poor quality material made by specific manufacturers, which raises the cost for everyone — even those with high quality processes.
Cell and gene therapy manufacturing would also benefit from greater standardisation. There is no central bank of assays that regulators have accepted and qualified. Companies are forced to find ones off the shelf or from the literature and hope they are accepted when they submit data to regulations. A regulator-industry consortium to share specific guidance on what assays are accepted, and what thresholds are required for release could help mitigate this issue.
When it comes to clinical trials, we should aim to make them both cheaper and faster. There is as of yet no substitute for human subjects, especially for the complex diseases that are the biggest killers of our time. The best model of a human is (still) a human.
Entering a trial as a patient can be a lengthy, bureaucratic process with onerous paperwork and consent requirements. Patient motivation to participate in a trial is often highest near when they first hear about the trial, and expires quickly. If logistical barriers and bureaucracy prevents them from enrolling in a timely manner they are likely to drop out of the process.
The GCP regulations that govern clinical trial conduct could be further adapted to be patient-centred and investigator-friendly. Unnecessary paperwork should be stripped down to focus core information. Burdensome consent forms that patients must review prior to enrolment should be made simpler, shorter, and easier for a layperson to understand, perhaps created in consultation with patient advocacy groups. Excessive safety reporting forms should be pared down to remove unnecessary information and standardised. Regulators can also help by being more explicit about what is required from companies running trials, so that companies do not overcompensate for ambiguity by providing excessive information.
We also need to make better use of the patients who enrol. This is especially problematic in rare diseases, where the supply of eligible trial volunteers may be lower than the amount needed to reach statistical significance.
Trials that would otherwise be conducted in isolation could be united under master ‘umbrella’ protocols. Umbrella trials test many drugs in parallel and use adaptive statistical methods to allocate patients to the arms with the most promising interventions13. This methodology was used to great effect in the pandemic with the RECOVERY trial that discovered dexamethasone’s efficacy against COVID-19. RECOVERY only cost around $25m for nearly 50,000 patients, or $500 per patient, far below industry standard. By stripping down the design to focus only on key metrics like survival, the protocol was straightforward to run across centres. Patients were identified from electronic health records, speeding up enrollment.
In practice, patient groups could work in partnership with industry to establish long-running platform trials for specific diseases. Patient input into development programs is beneficial in general, and often speeds up overall timelines. Patients can help identify issues that lead to delays or refusals to participate, like removing unpleasant or burdensome tests. Consider certain rare diseases like cystic fibrosis which have built strong communities and disease registries around specific diseases. Strong patient communities can quickly identify patients for trials — ideally these active networks could be expanded beyond highly-engaged rare disease communities.
Incentivising uneconomical, yet desirable, development
Lastly, we should aim to make sure that new and potentially valuable technologies get the support they need to mature, instead of withering on the vine.
Biotech is an inherently capital intensive industry, and so underinvestment leads to waste. When money is tight, investors and biotechs may underinvest in development programs or spread their investments too thin, requiring corners to be cut in manufacturing or trial design. Underinvestment manifests as trials that are too small to properly provide actionable information, or manufacturing that skimps on proper process and controls leading to development delays or discontinuations down the line. It is often preferable to do fewer things better, and underinvesting means we don’t learn enough from each dollar we spend.
A potential driving force behind underinvestment has been the trend towards greater fragmentation of R&D activity and investment. The past few decades have seen large pharmaceutical companies deprioritise investments in internal early-stage drug discovery in favour of an externalised model where drugs and technologies are acquired from smaller venture-backed biotechs. This externally-focused model has delivered many successes, and venture capitalists are undoubtedly vital contributors to the biotech ecosystem. However, venture capitalists have a different set of incentives to those of a large company operating a well-funded industrial research lab. Abdicating control over early stage capital allocation to venture capitalists may have led to the under-resourcing of speculative capital-intensive applied research in a manner that echoes the fall of large industrial R&D labs like Xerox’s PARC or AT&T’s Bell Labs.
Underinvestment and short-term thinking is particularly regrettable when it comes to immature, but promising, technologies like cell and gene therapies. During the pandemic, there was a speculative bubble in gene therapy and gene editing firms which has since burst. While many of these firms came to market with high hopes, it eventually became clear that these technologies were not yet mature enough to live up to lofty investor expectations. Stock valuations of gene therapy and editing biotechs have come down by as much as 80%, many have made deep cuts to their workforce and development programs, or ceased operations entirely. The FDA has itself even expressed concern about the future commercial viability of cell and gene therapies. By spreading out the capital and expertise among many firms, and handing the reins to short-term focused investors, we may have inadvertently engineered a scenario in which no one firm allocates enough resources and time to sufficiently invest in developing the fundamental technology.
ARPA-H or similar early stage funding institutions might be well placed to step in and provide this long-term vision and support to help early stage ATMPs get out of the lab and into the clinic. For newer types of drugs like gene therapies, the best methods of manufacturing and ensuring quality are still being established. For instance, centralised compliant manufacturing facilities for complex cell and gene therapies could enable small companies to make batches to test a therapeutic hypothesis, before having to invest considerable resources into independent process development and manufacturing scale up. Few biotechs today have the resources to change their manufacturing process after getting into patients, because the extensive comparability studies required are too expensive. A gene therapy developer can get locked into a suboptimal (e.g. difficult to scale) process before they’ve even dosed their first patient. Ideally, modest changes in the manufacturing process for development-stage gene and cell therapies should not require a full expensive comparability study to address. Further, instrumentation for assays is expensive and underdeveloped in cell and gene therapy — better tools allowing for multiplexed assays would help lower costs.
Expanding and centralising investment in capabilities that could be shared by innovators in the field could help reduce redundant expenditure and drive these next generation technologies down the cost curve.
Coda
Fundamentally, the way to recapture the biopharmaceutical industry’s lost productivity is to make experimentation cheaper. Rapid iterative trial-and-error of the sort employed by Janssen in his research is a key driver of progress. As was the case with haloperidol, many of our most impactful drugs were serendipitous discoveries. When experimentation is cheap, companies are likely to be open to financing more exploratory research. On the other hand, expensive development and marginal returns engenders an accounting-based mindset that reduces upside risk in favour of safe incremental plays. The less we explore the less we will find.
Janssen pharmacetica is now a large multinational with 40,000 employees, and part of Johnson & Johnson. As Janssen’s company has changed, so has the industry. Shortly before his death in 2003, Janssen reflected on how the rising costs of drug development had constrained his company and changed the nature of the industry. Such costs are a shame, he said:
Especially for people with a serious illness not common enough to make it cost effective for the pharmaceutical industry to invest in finding a cure. I am 75 years old, so old enough to get angry about this. Especially when I think of all the drugs that are not being developed.
Reversing this long term decline in R&D efficiency is the grand challenge of the biopharmaceutical industry. If we continue to simply place policy pressure on drug prices without a concomitant decrease in development cost or failure rates, biopharmaceutical progress will suffer as investors pull out — after all, there are easier ways to make money.
-
It’s reasonable to ask how it’s even possible to find useful drug molecules at all given the vastness of chemical space (over 10^60 possible drug molecules). One possible answer is that nature, through evolution, has chanced upon many bioactive molecules, and we are mostly building off that inheritance. In support of this are data showing that the screening libraries that we use as the starting point for new drugs are biassed towards drug molecules with natural origins ↩
-
There can be more than one primary endpoint. There are also secondary endpoints, which are usually outcomes that the trial is too small or short to see a statistically significant difference on (such as overall survival), or are nice-to-have, such as quality of life questionnaires ↩
-
In reality volunteers get ‘best supportive care’, which includes treatments to manage symptoms. Placebos, when used as a comparator, are often given on top of a background of standard of care therapies. Selecting an appropriate comparator for a trial can be a complex question. For one, standards of care may differ between countries and centres. Choosing the wrong comparator can wreck the prospects of a new drug, as regulators, physicians and payers expect to see data on how new drugs compare to drugs in actual use ↩
-
For example, inadvertently encouraging participants in the active drug arm to exert more effort in a functional test. The 6-minute walk test is one such functional test that is often used as a measure of endurance, and is highly sensitive to how hard participants try ↩
-
Regulators may conduct preapproval inspections of manufacturing facilities to ensure that the actual processes for manufacturing, analysis and stability testing are consistent with the ones described in the application for approval. They will also audit data, check that the facility is actually capable of scalable manufacturing based on the equipment in place, and check for contaminants ↩
-
Clinical trials for a given drug don’t necessarily stop after approval: companies often need to run post-marketing trials that follow patients for years to confirm if efficacy and safety of the drug holds up in the real world. Additionally, if companies want to advertise their drugs for new indications they will have to repeat the process for each additional patient type, although they may be able to skip phase I ↩
-
Incidentally this vaccine was produced by Johnson and Johnson’s Janssen unit, the same company that Paul Janssen founded. It should be mentioned that the Johnson and Johnson vaccine overall had a positive risk-benefit ratio, but the blood clot risk contributed to a more favourable risk-benefit ratio for the mRNA vaccines, so mRNA vaccines were preferentially recommended by the ACIP ↩
-
For an example of different standards applied to newer versus old drugs, see the history of rofecoxib ↩
-
There are precedents of safety events in trials leading to long delays in research. The death of Jesse Gelsinger, the first in a gene therapy trial, led the FDA to halt gene therapy trials in the US and mostly set the field back by a decade. There is still no licensed vaccine for group A Streptococcus infections, in part because the FDA banned “Group A Streptococcus organisms or their derivatives” from vaccines in 1976 after a small uncontrolled trial identified potential safety risks, effectively stopping research. However, this ruling was revoked in 2006. While situations like Gelsinger’s are tragedies and extreme care should be taken not to repeat the mistakes that led to his death, overextrapolation from isolated cases to the broader field is damaging for progress ↩
-
Of course, it could also be that the industry is delivering the same or more value, but capturing less of that value (increasing consumer surplus). Alternatively, more expensive manufacturing or policy constraints could raise costs and reduce profits in a way that is independent of value created ↩
-
While a useful indicator, Eroom’s law is flawed in some important ways that are worth mentioning. First, it doesn’t capture an important output of research, which are indication expansions of already approved drugs. Many of today’s leading drugs are approved for multiple indications. For instance, Humira, a top-selling autoimmune disease drug, is FDA approved for 9 separate indications.Second, Eroom’s law mixes the effects of fewer successes with rising costs without giving a clear picture of which factor is driving the trend ↩
-
Lest I be accused of claiming otherwise, I should note that the decreases in cancer and cardiovascular death rates are not solely attributable to pharmaceuticals. Lifestyle changes, such as smoking cessation, have also played an important role ↩
-
Umbrella trials could also be a useful mechanism for relatively cheap drug repurposing — finding new uses for old off-patent drugs – as industry is not necessarily incentivised to run repurposing trials on a stand-alone basis ↩